NAD+ 100mg
$48.00
- Buy 5 and save 10%
- Buy 10 and save 15%
Legal Notice Regarding This Product
THIS PRODUCT IS STRICTLY INTENDED FOR RESEARCH USE ONLY. It is designated solely as a research chemical. Usage is confined to in vitro testing and laboratory experimentation exclusively. Information available on our website about this product is for educational purposes only and should not be interpreted as an endorsement or directive for use outside these bounds. Introducing this product into humans or animals by any route is strictly prohibited by law. Handling of this product should be undertaken only by licensed, qualified professionals.
This product is not to be used as a drug, food, or cosmetic. Misbranding, misusing, or mislabeling this product as a drug, food, or cosmetic is illegal. Users are responsible for complying with all applicable laws and regulations regarding the handling and use of this product.
THE PRODUCTS PURCHASED ON THIS WEBSITE ARE INTENDED FOR RESEARCH CHEMICAL USE ONLY. These products are not intended to be used for human or animal consumption and/or ingestion of any kind. These products should not be used as food additives, drugs or household chemicals. These products should only be used by Qualified Professionals.
Bodily introduction into Human and Animals of any kind is strictly forbidden by law.
All the product information on this website is for educational purpose only.
NAD+
Product Usage: THE PRODUCT IS INTENDED AS A RESEARCH PURPOSE ONLY. This designation allows the use of research chemical strictly for in vitro testing and laboratory experimentation only. All product information available on www.peptidesworld.com is for educational purposes only. Bodily introduction of any food, cosmetic and may not be misbranded, misused or mislabeled as a drug, food or cosmetics.
Highlights:
Therapeutic potential of boosting NAD+in aging and age-related diseases.
- Therapeutic potential of boosting NAD+ in aging and age-related diseases.
- Diabetes, improvements in glucose tolerance.
- Non-alcoholic fatty liver disease.
- Atherosclerosis.
- Supplementation with the NAD+ intermediate NMN was able to restore retinal function.
- Depression.
- Protection against neurodegeneration and DNA damage.
Therapeutic potential of boosting NAD+ in aging and age-related diseases
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor in all living cells that is involved in fundamental biological processes. NAD+ depletion has been associated with hallmarks of aging and may underlie a wide-range of age-related diseases, such as metabolic disorders, cancer and neurodegenerative diseases. Emerging evidence implicates that elevation of NAD+ levels may slow or even reverse the aspects of aging and also delay the progression of age-related diseases. Here we discuss the roles of NAD+-synthesizing and -consuming enzymes in relationships to aging and major age-related diseases. Specifically, we highlight the contribution of NAD+ depletion to aging and evaluate how boosting NAD+ levels may emerge as a promising therapeutic strategy to counter aging-associated pathologies and/or accelerated aging. [1]
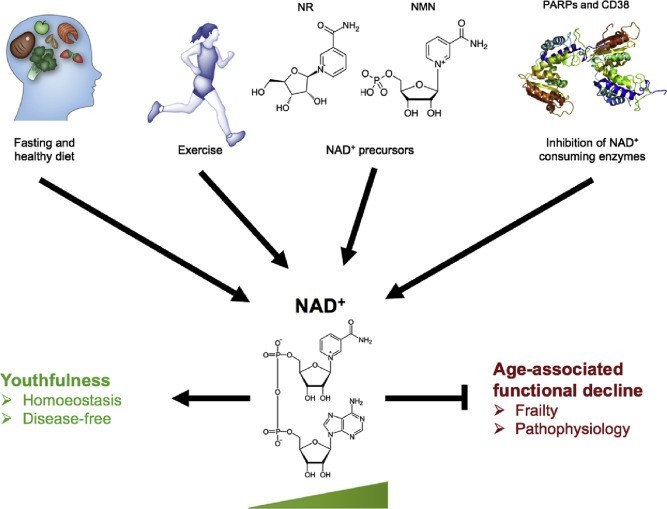
Fig. 2. Physiological and pharmacological strategies for boosting NAD+ levels. Inhibiting the age-related decline in NAD+ levels is critical for preventing age- or disease-related frailties. Physiological strategies that could potentially boost NAD+ levels include exercise, fasting, and maintaining a healthy diet. Pharmacologically, boosting NAD+ can be achieved via either supplementation of its precursors, NR and NMN, or inhibition of its consumers by use of CD38and PARPs inhibitors. Figure modified from Fang et al. 2017 and Fivenson et al. 2017 [1].
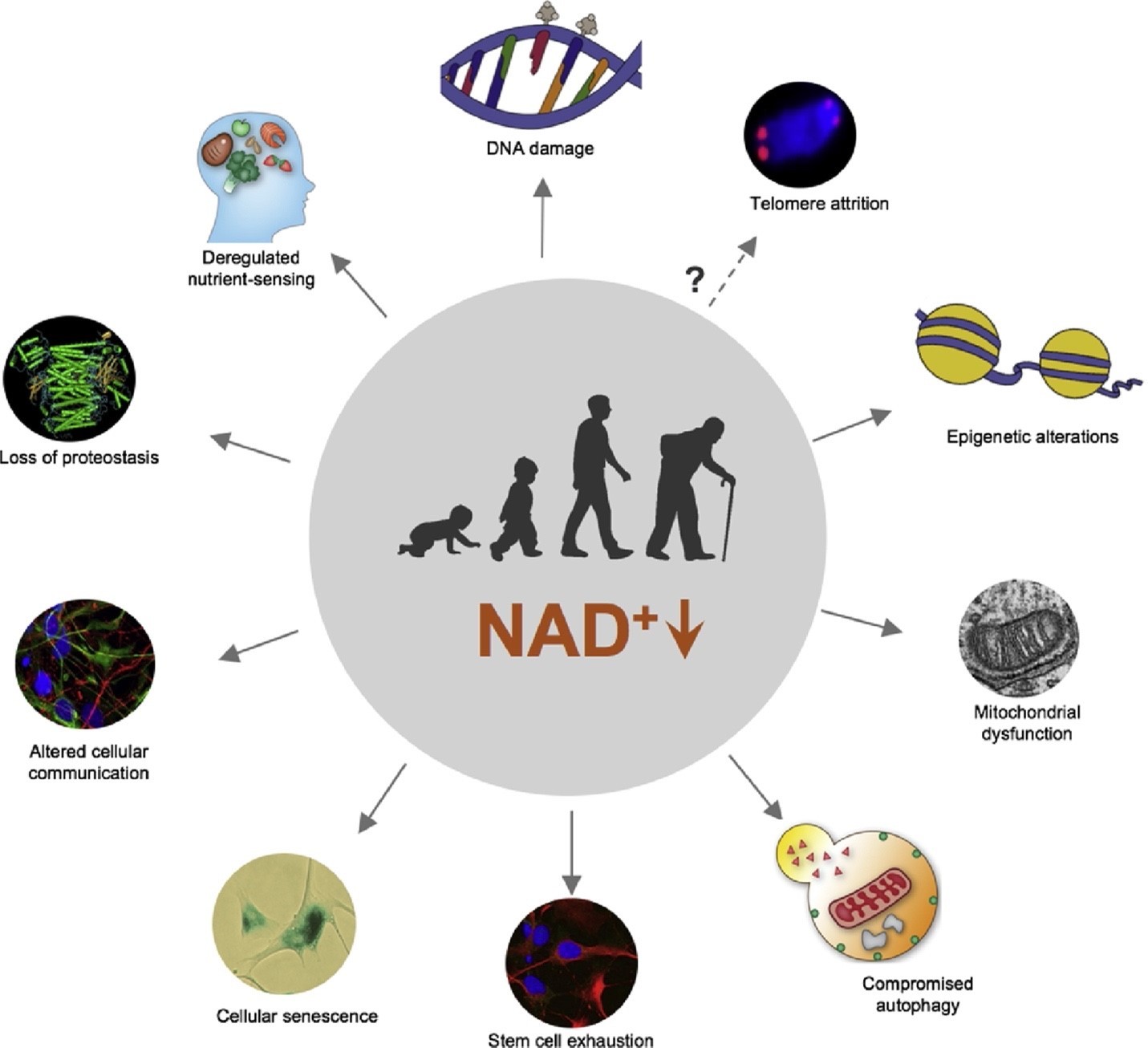
Fig. 1. NAD+ decline at the core of hallmarks of aging. A schematic representation of age-dependent decline in NAD+ levels which contribute to ten hallmarks of aging, namely DNA damage, epigenetic alteration, deregulated nutrient-sensing, loss of proteostasis, altered cellular communication, cellular senescence, stem cell exhaustion, mitochondrial dysfunction, compromised autophagy, and possibly telomere attrition. Figure modified from Fang et al. 2017 [1].
1. What is NAD+
Nicotinamide adenine dinucleotide (NAD+) is an important cofactor in all living cells that is involved in fundamental biological processes, namely metabolism, cell signalling, gene expression, DNA repair, among others . Originally, Harden and Young described NAD+ in 1906 as a molecular fraction (“cozymase”) that accelerated fermentation in yeast extracts. Over subsequent years, NAD+ was identified as a nucleoside sugar phosphate, which plays a role in redox reactions. However, evidence stemming from recent studies have unveiled numerous roles of NAD+ metabolism on aging and longevity. In particular, an age-dependent decline in NAD+ levels have consistently been reported, possibly due to an imbalance in the synthesis and consumption of NAD+. Decreased levels of NAD+ are associated with the hallmarks of aging as well as several age-related diseases, including metabolic disorders, cancer and neurodegenerative diseases. Replenishment of NAD+ levels via administration of its precursors have been demonstrated to display beneficial effects against aging and age-related diseases. Importantly, boosting NAD+ levels have been shown to extend lifespan of various laboratory animal models including worms, flies, and rodents. [1]
As a cofactor, NAD+ is found in abundance in the mitochondria, cytoplasm, and nucleus. It is essential for many cellular metabolism pathways that include: glycolysis, fatty acid β-oxidation, and the tricarboxylic acid cycle. Whilst the reduced form of NAD+ (NADH) is a primary hydride donor in the production of ATP via anaerobic glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) [1]. On the other hand, NAD+ is consumed by the NAD+-dependent sirtuins and the DNA damage sensors poly (ADP-ribose) polymerases (PARPs) in the processes of protein deacetylation and poly-ADP-ribosylation (PARylation), respectively. In addition, NAD+ glycohydrolases (i.e. CD38 and CD157) also consume NAD+ via conversion of NAD+ into ADP-ribose (ADPR) or cyclic-ADPR [1]. Thus, the importance of NAD+ has expanded from a key element in intermediate metabolism to a critical regulator of multiple cell signalling pathways; and is now a major player contributing to aging and age-related diseases.
In mammals, NAD+ is synthetized from a variety of dietary sources, including NAD+itself (it is metabolized in the gut, then synthesized again in cells) as well as from one or more of its major precursors that include: tryptophan (Trp), nicotinic acid(NA), nicotinamide riboside (NR), nicotinamide mononucleotide (NMN), and nicotinamide (NAM). Based upon the bioavailability of its precursors, there are three pathways for the synthesis of NAD+ in cells: (i) from Trp by the de novobiosynthesis pathway or kynurenine pathway; (ii) from NA in the Preiss–Handler pathway; and (iii) from NAM, NR, and NMN in the salvage pathway.
Accumulating evidence demonstrates an age-dependent decline in NAD+ levels and associate its depletion to several hallmarks of aging and age-related diseases (Fig. 1) . Here, we summarize the roles of NAD+-synthesizing and -consuming enzymes in aging and age-related diseases. Specifically, we highlight the contribution of NAD+ depletion to mammalian aging and evaluate how boosting endogenous NAD+levels might emerge as a promising therapeutic strategy to counter aging-associated pathologies and/or accelerated aging. [1]
Recent progress on the roles of NAD+ in aging
Mounting evidence has indicated that NAD+ levels decline with age in multiple types of tissues, which include the liver, skeletal muscle, adipose tissue, heart, brain, kidney, pancreas, lungs, spleen, skin, as well as extracellular fluids. In addition, an age-dependent decline in NAD+ levels in Caenorhabditis elegans (C. elegans), mice, and human post-mortem tissues are reported. Thus, highlighting a universal age-dependent decrease of NAD+ across species. However, it remains elusive whether this is due to increased NAD+ consumption and/or decreased synthesis. In this section, we provide an overview of methods that could potentially boost endogenous levels of NAD+ [1].
2. Recent progress on the roles of NAD+ in aging
Mounting evidence has indicated that NAD+ levels decline with age in multiple types of tissues, which include the liver, skeletal muscle, adipose tissue, heart, brain, kidney, pancreas, lungs, spleen, skin, as well as extracellular fluids . In addition, an age-dependent decline in NAD+ levels in Caenorhabditis elegans (C. elegans), mice, and human post-mortem tissues are reported. Thus, highlighting a universal age-dependent decrease of NAD+ across species. However, it remains elusive whether this is due to increased NAD+ consumption and/or decreased synthesis. In this section, we provide an overview of methods that could potentially boost endogenous levels of NAD+ [1].
2.1. NAD+ and aging
NMN is a physically stable natural compound that serves as an efficient NAD+precursor. In mammals, NMN is synthesized from nicotinamide, a form of water-soluble vitamin B3 and 5′-phosphoribosyl-1-pyrophosphate (PRPP), by the rate-limiting enzyme, nicotinamide phosphoribosyl transferase (NAMPT). In addition, it can also be synthesized from NR via NR kinases (NRKs)-mediated phosphorylation reactions. NMN is subsequently converted into NAD+ by NMN adenylyl transferases(NMNATs). Over the years, it has become increasingly evident that systemic administration of NMN in rodents enhances the biosynthesis of NAD+ in various peripheral tissues including liver, pancreas, adipose tissue, heart, skeletal muscle, kidney, eyes, and blood vessels. Furthermore, NMN has also been shown to elevate levels of NAD+ in hypothalamusand hippocampus following an intraperitoneal injection, thereby indicating its ability to penetrate the blood-brain barrier (BBB). More importantly, long-term (1-year) oral administration of NMN (up to 300 mg/kg) has recently been shown to be well tolerated without any obvious deleterious or toxic effects in normal wild type C57BL/6 mice.
NMN has been shown to have remarkable beneficial effects that counter normal aging. In models of aging, long-term administration of NMN protects against age-associated functional decline as demonstrated by increases in energy metabolism, insulin sensitivity, lipid metabolism,
mitochondrial oxidative metabolism, eye function, bone density and immune function. On the other hand, it suppressed age-related changes in gene expression and adipose tissue inflammation. Moreover, it has been shown to maintain neural stem/progenitor cell population and restore skeletal muscle mitochondrial function as well as arterial function in aged mice. In addition, loss of enzymes involved in NAD+ synthesis, namely NAMPT during the process of aging, led to decrease in NAD+ content and reduced SIRT1 activity; consequently, promoted cellular senescence in retinal pigment epithelium, which performs numerous functions critical to retinal health and visual function [1].
NMN administration may counteract age-predisposed metabolic diseases and neurodegeneration. In age-related pathophysiological conditions, NMN ameliorated impairments in glucose tolerance and promoted insulin secretion/sensitivity in age- or diet-induced diabetic mice, Nampt+/− mice, as well in aged wild-type and β cell-specific Sirt1-overexpressing (BESTO) mice. Likewise, promotion and overexpression of the mitochondrial Nmnat3 in mice, also involved in NAD+ biosynthesis, resulted in improved glucose tolerance during the process of aging as well as in models of high-fat induced obesity. The beneficial effect was suggested to be a result of improved mitochondrial function and by an independent mechanism of NAD+–SIRT1–PGC1α axis, which despite previously being reported to contribute to improved mitochondrial function, was not activated in these transgenic mice despite elevated levels of NAD+. Furthermore, NMN protects the heart and brain from ischaemia-induced damage. In rodent models of Alzheimer’s disease (AD), administration of NMN decreased AD-associated β-amyloid (Aβ) pathology and improved cognitive function. In addition, it restored mitochondrial function and ameliorated inflammation, synaptic loss as well as protected against neuronal cell death. The beneficial effect of NMN was also evident in premature aging conditions as demonstrated by extended lifespan and improved healthspan in the C. elegans model of xeroderma pigmentosum group A (XPA, a nucleotide excision DNA repair (NER) disorder with severe neurodegeneration), and Ataxia telangiectasia (A-T, due to mutation of ATM which encodes a master regulator of DNA damage response). Moreover, in mice with hypomorphic BubR1 (a mitotic check-point kinase) exhibited characteristics of premature aging as evidenced by shorter lifespan, which was restored by NMN supplementation. Altogether, these findings highlight NMN induced restoration of NAD+ levels serve as beneficial therapeutic strategy in countering aging as well as age-related pathological conditions in animal models.
2.2. NR and aging
NR is a natural NAD+ precursor. It can directly be converted into NMN via the activity of NRKs, thereby bypassing the requirement of the NAMPT in the salvage pathway, and therefore offers to provide an additional pathway for elevation of NAD+ levels. Similar to NMN, NR also exhibited beneficial effects in protection against aging and age-related diseases. It has been shown to promote longevity as well improve healthspan in multiple laboratory animal models. In age-related disease, in particular obesity, diabetes and cardiovascular conditions NR was able to decrease weight gain, improve glucose tolerance and increase survival rates, respectively in rodents. In addition, in models of diabetes and high-fat diet, NR was able to improve metabolic function and reduce lipid accumulation as well as increasing lifespan. Furthermore, supplementation of NR reversed the progressive wasting syndrome and restored endurance in Nampt skeletal muscle knockout mice, mdx model of Duchenne’s muscular dystrophy [1].
NR ameliorates neurodegeneration in animal models. In animal models of age-related neurodegenerative diseases such as AD and Parkinson’s disease (PD), NR has been shown to improve memory, learning, motor function and mitochondrial function as well as protected against neuronal cell death. In particular, chronic NR administration in the amyloidogenic models of AD delayed the development and progression of Aβ pathology in AD mice, SH-SY5Y cells, and AD C. elegans. In addition, it was able to promote longevity and inhibit/delay cognitive decline in AD C. elegans and mice, with the enhanced process of mitochondrial proteostasis and modulation of β-secretase 1 (BACE-1) activity via peroxisome proliferator-activated receptor-gamma coactivator 1 (PGC)-1alpha highlighted as possible underlying mechanisms. Further evidence reinforcing the beneficial effects of NR as a therapeutic strategy in AD was provided by a recent study using triple transgenic model of AD (3 × Tg), which exhibited not only reduced phosphorylated tau pathology and inhibited cognitive decline; but also, normalised AD-associated neuroinflammation and synaptic dysfunction. Moreover, DNA damage was reduced, in addition, following chronic administration of NR in a DNA repair-deficient 3 × Tg/Polβ +/− mouse model. The underlying mechanism proposed was the reduction in DNA damage results in reduced activity of NAD+ consuming enzyme PARPs that is involved in DNA repair; thereby increase in the levels of NAD+, which in turn contributes to neurogenesis and inhibits AD-associated pathology, neuroinflammation and mitochondrial dysfunction. Likewise, NR treated induced pluripotent stem cells (iPSCs) derived from PD patients harbouring mutations in the lysosomal enzyme β-Glucocerebrosidase (GBA) gene (GBA-PD), the most common genetic risk for PD, resulted in elevated levels of NAD+ and NAM which coincided with improved mitochondrial morphology and function. Mitophagy was suggested to be a possible mechanism promoted by NR, which may underlie improved mitochondrial quality control. In addition, flies model of GBA-PD expressing human N370S GBA raised on food containing NR displayed improved motor function and significantly decline in loss of dopamine-containing neuronal population. Additionally, NR has shown significant neuroprotection in a series of DNA repair-deficient premature aging diseases, including XPA, A-T, and Cockayne syndrome (CS, due to impairment of NER) [1].
2.3. NAM and aging
NAM is also a precursor for NAD+ and a key molecule involved in energy metabolism. Low doses of NAM have been shown to increase lifespan in yeast and C. elegans, however, higher doses have been associated with reduced lifespan via inhibition of Sir2 activity. In models of aging and high fat diet-induced obesity, NAM improved healthspan although it failed to extend lifespan as illustrated by comparable mean and maximum lifespan. In the model of obesity, it has able to restore glucagon storage to similar levels as age-matched standard-diet mice as well as ameliorate diet-induced hepatosteatosis, oxidative stress and inflammation. It was suggested that the beneficial impact of NAM may be attributes to improved mitochondrial function and countering age and high fat diet induced DNA damage. Further evidence reinforcing the beneficial impact of NAM stemmed from a mouse model of glaucoma, which inhibited the development glaucoma in the eyes. Moreover, these findings were replicated by Nmnat1 gene therapy whereby an intravitreal administration of adeno-associated virus AAV2.2 carrying a plasmid to overexpress murine Nmnat1 under a CMV promoter was performed in D2 eyes. The improvement of mitochondrial health and metabolism was suggested to be the underlying mechanism for countering glaucoma mediated by NAM supplementation and Nmnat1 gene therapy. [1]
Neurodegeneration and DNA damage
PARP inhibition
The NAD+ consuming enzymes, PARPs, cleave NAD+ into NAM and ADP-ribose(ADPR), as a result generating a chain of ADPR. PARP1 is the most abundant PARPs, which is ubiquitously expressed and is a major consumer of NAD+ in response to DNA damage whereby it contributes to facilitation of the DNA repair process. Both PARPs and sirtuins share NAD+ as a common substrate thus compete for its consumption. It has been reported that PARP1 activity increased with the inevitable process of aging possibly due accumulation of DNA damage. Consequently, the NAD+ pool is depleted which results in reduced activity of sirtuins. Evidence stemming from genetic deletion of PARP1 in mice as well as pharmacological inhibition of PARPs revealed increase in NAD+ content and enhanced activities of sirtuins, in particular SIRT1 and SIRT6. Elevated SIRT1 activity was associated with increased mitochondrial content and oxidative metabolism as well as protection against metabolic dysfunction, DNA damage, and neurodegeneration. [1]
In addition, increased PARP1 activity has been reported in animal models of age-related neurodegenerative diseases, namely AD and PD. Deletion of PARP1 in AD mice protected against cognitive decline as well as attenuated neuroinflammation and Aβ-induced neurotoxicity . In PD rodents, PARP1 pharmacological inhibitors or deletion resulted in resistance to the toxic effects and loss of dopamine-containing neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA), respectively. Thus, implicating the contribution of PARP1 hyperactivity in processes leading to neurodegeneration. Moreover, hyperactivity of PARP1 is reported in models of premature aging that resulted decrease in SIRT1 activity, a feature that was reversed by replenishment of NAD+ using its precursors. Therefore, a potential therapeutic counter for depleted NAD+ pool in aging and age-related diseases could be inhibition of its consumer, PARPs, which will enable activity of sirtuins that plays a pivotal role in regulating cellular processes.
3.2 CD38 inhibition
CD38 is one of the primary NADases in mammals. It can modulate the levels of NAD+via hydrolysis of NAD+ itself as well degradation of its precursors, NMN and NR. Primarily, CD38 hydrolyses NAD+ to produce of ADPR and NAM. It is a key player in several physiological processes, including nuclear Ca2+ homoeostasis, immunity, inflammation, glucose and lipid homoeostasis, transferring of mitochondria from astrocytes to neurons, as well as social behaviour. An age-dependent increase in levels of CD38 protein has been reported in multiple tissues and organs, which as a result contributes to NAD+ decline. Therefore, CD38-dependent modulation of NAD+ can alter the activity of NAD+-consuming enzymes and affect cellular signalling and metabolism. Inhibition of CD38 can also promote NAD+ levels and improve glucose and lipid metabolism, which protects against age- and diet-induced diabetes and obesity. In APP/PS1 model of AD, CD38 depletion resulted in elevation of NAD+ levels that were associated with decrease in Aβ pathology and associated neuroinflammation accompanied by improvement spatial learning behaviour. However, due to the reported important neuroprotective activities of CD38, further stringent and comprehensive evaluation of the procedures of CD38 inhibition as a safe anti-aging strategy.
3.3 NNMT knockdown
Nicotinamide N-methyltransferase (NNMT) catalyses the methylation of NAM N1-methyl-2- pyridone-5-carboxamide (2py) and N1-methyl-4-pyridone-3-carboxamide (4py) using the universal methyl donor S-adenosyl methionine (Met) (SAM). Both products of NAM methylation are eventually excreted in the urine; thus, NNMT removes NAM from the NAD+ biosynthesis pathway and thereby contributes to decrease in levels of NAD+ [80]. It is predominantly expressed in the liver and adipose tissue but is also found in other tissues including kidney, lung, muscle, heart, brain, and tumour cells. NNMT has been shown to be involved in various disease conditions such as metabolic disorders, neurodegenerative diseases and cancer. In conditions such as obesity and diabetes, NNMT levels have been reported to be upregulated significantly, which in turn are associated with the disease phenotype. In fact, genetic knockdown as well as pharmacological inhibition of NNMT was shown to be beneficial in protection against obesity in rodent models of obesity.
3.4. Genetic promotion of NAD+ biosynthesis
In addition to abovementioned techniques that have shown to enhance NAD+biosynthesis, genetic tools in the form of Lactobacillus brevis (LbNOX), a water-forming NADH oxidase, has been demonstrated to also induce an increase in compartment-specific levels of NAD+/NADH ratio in HeLa cells. It catalyses a four-electron reduction of oxygen to water (2 NADH + 2H+ + O2 → 2 NAD+ + 2 H2O). The NAD+/NADH ratio plays a role in cellular metabolism by affecting the activity of NAD+-dependent enzymes such as sirtuins. LbNOX was shown to ameliorate proliferative and metabolic defects induced by a dysfunctional electron transport chain (ETC) by recycling the pool of NAD+. This, therefore offers to be a novel approach whereby NAD+ levels could potentially be boosted via genetic manipulation in order to understand the fundamental molecular mechanisms in models of agingand age-related diseases.
4. Methods to detect subcellular NAD+
In view of the importance of NAD+ in life, aging, and diseases, it is necessary to accurately detect subcellular NAD+ levels to further unveil its intracellular functions as well as to develop sub-cellular organelle-targeted therapeutic approaches. Traditionally, several assays, such as high-performance liquid chromatography(HPLC)-based methods (e.g., HPLC/MALDI/MS) and fluorometric-based commercial kits have been utilised to detect whole NAD+ at both cellular levels and/or sub-cellular levels (by isolating sub-cellular fractions ahead). There have some challenges of using these methods to accurately detect sub-cellular NAD+ levels because of the highly instability of NAD+ as well as impossibility of NAD+ detection in live cells/tissues. The recent development of genetically encoded fluorescent biosensors such as SoNar and a biosensor with a bipartite NAD+-binding domain have enabled imaging of relative levels of free NAD+ in the subcellular compartments. Quantification of NAD+ using fluorescent biosensor targeted to different compartments of mammalian cells showed that mitochondria contain more than twice as much free NAD+ as other compartments. Specifically, the concentration of NAD+ was reported to be approximately 110 μM in the cytoplasm and the nucleus relative to 230 μM in the mitochondrion. These levels are consistent with other reports demonstrating that, in highly metabolically active, post mitotic cells, such as neurons, mitochondria have higher NAD+ levels compared with other sub-cellular compartments. It was further demonstrated genetic and pharmacologic inhibition of NAMPT result in a reduction in NAD+ concentration in all compartments, but depletion of mitochondrial NAD+ occurred at a slower rate. Furthermore, the nuclear and cytoplasmic pools were shown to be readily exchangeable, whilst the mitochondrial pool may maintain mitochondrial NAD+ levels via NAD+ biogenesis by mitochondrial isoform NMNAT3 and import from the cytoplasm. Another recent development has been offered in the form of NAD+ flux quantification that is isotope-tagged and used for analysis of NAD+ metabolism. It demonstrated that approximately 50% decrease in NAD+ consumption following treatment with SIRT1/2 and PARP1/2 inhibitors; thereby implicating both are major consumers of NAD+. The use of such approaches in models of aging and age-related diseases in combination with NAD+ promoting methods would allow the identification of subcellular localisation as well as consumption, which in turn would allow to identify the underlying molecular mechanisms and pathways attributed to NAD+ benefits [1].
5. Clinical translation
Encouraged by significant and replicable benefits of NAD+ precursors, NR and NMN, in aging and disease animal models, a series of clinical trials of NR and NMN in normal aged population and individuals with diseases have been performed. NAD+ precursors, in particular NR has been demonstrated to elevate blood concentration of NAD+ in healthy individuals in a dose-dependent manner and without any toxic effects. In particular, single oral self-administration of NR (1000 mg) over a period of seven days in a 52 years old male increased blood concentration of NAD+ by 2.7-folds and 45.5-fold increase in nicotinic acid adeninedinucleotide (NAAD), an NAD+ biosynthesis intermediate. In addition, a randomized double-blind pharmacokinetic study of single oral administration NR (doses: 100 mg, 300 mg, and 1000 mg) with seven-days gap conducted in 12 healthy patients (aged 30–55 years old) revealed a dose-dependent increase in NAD+ and NAAD levels, with no reported adverse effects. The encouraging results in animal models of aging and age-related diseases of chronic administration of NAD+ precursors have led to studies in humans. An eight-week randomized, double-blinded, placebo-controlled study in 120 healthy adults (60–80 years old) demonstrated NR (250 mg and 500 mg) induced dose-dependent increase of blood NAD+ level that becomes apparent after 4-weeks and is sustained till the end of the study. Importantly, no serious adverse effects were reported, thereby implicating the chronic administration of NR is a safe and effective way to increase NAD+ levels. These findings are reinforced by a 2 × 6-week randomized, double-blind, placebo-controlled crossover clinical trial conducted in 55–79 years old individuals that showed NR (oral 500 mg, twice a day) to be well tolerated and able to effectively elevates NAD+ levels in healthy adults. Moreover, it was able to reduce systolic blood pressure and aortic stiffness, which are considered measures of cardiovascular disease. Thus, not only the NAD+ precursors are safely administrated but they may also recapitulate the beneficial effects that were evident in animal models, which is an exciting prospect for future clinical trials. However, in conditions such as pancreatic cancer, cell growth has been shown to be dependent of the NAD+ salvage pathway. Hence, inhibition of NAD+ synthesis (via Nampt inhibition) and/or promotion of its consumption (via CD38 NADase) prevented cancer cell growth. Therefore, implicating that there should be a thorough evaluation for the use of approaches that promote NAD+biosynthesis as it may be contributor rather than a counter-mechanism in certain conditions. Clinical trials of NAD+ precursors on age-related diseases, such as diabetes, premature aging diseases, and neurodegenerative diseases are in progress.
5.1 Outstanding questions and future perspectives
Age is the primary cause of the majorly of human diseases and interventional strategies/therapeutics targeting on human aging is arguably the most efficient approach to achieve healthy aging and the improvement of the quality of life worldwide. Although, maintaining a healthy diet, fasting, and exercise may improve the quality of life, it may not be feasible option for all individuals. Therefore, the beneficial effects of NAD+ discussed in the present review, highlight possible ways for improving the quality of life via hindering numerous pathological hallmarks of aging and thereby improving the quality of life and delay age-related diseases (summarized in Fig. 2). Preclinical evidence of NAD+ replenishment that could potentially delay and/or prevent metabolic conditions, hearing loss, muscle atrophy, and cognitive decline are really encouraging for future perspectives. Moreover, NAD+ precursors, in particular NR has been shown to be safely administrated and also able to demonstrate improvement of cardiovascular functions in human. Thus, implicating a possible translational aspect of preclinical benefits of NAD+ supplementation, which is an exciting prospect and opens avenues for future studies to test the impact of elevated NAD+ biosynthesis in aging and age-associated diseases in human.
Despite extensive research on NAD+ biosynthesis and its implications in health and disease, there are major questions that are yet to be explored. Firstly, what levels of NAD+ are to be associated with healthy aging and age-related diseases? In particular, it is of great relevance to elucidate organ and sub-cellular localisation as well levels of NAD+ in health and disease. Such observations would allow to map health- and disease-specific alterations of NAD+, which could be utilised to develop therapeutic interventions that promote NAD+ in a region-specific manner as a counter-mechanism. Decline in NAD+ has been implicated during the process of aging and age-associated diseases, thereby may be affect various processes that are likely to key contributors and drivers of associated dysfunction. Thus, preclinical studies driven towards unveiling the pathways and mechanisms underlying the beneficial effects of NAD+ replenishment in healthy aging and models of disease are required in order to understand how NAD+ contributes to delay and/or prevent hallmarks associated with aging. In particular, it is important to elucidate whether individual or multiple hallmark(s) mechanisms associated with aging are countered by NAD+ replenishment. This would allow understanding of the mode of NAD+ action and the interconnection and contribution of the various hallmarks of aging. NAD+ precursors have thus far shown to be safely and effectively administrated in healthy old humans, elevating NAD+ levels in blood, but its safety and tolerance is yet to be determined in individuals with age-related diseases. Therefore, future clinical trials are required to assess the safety of NAD+ precursors in patients with age-associated diseases such as diabetes and AD. Though, as abovementioned, careful evaluation of the role of NAD+, whether friend or foe in disease, must be taken into account for each disease-condition. Altogether, NAD+ replenishment may serve as a potential therapeutic strategy for aging and multiple conditions to improve the quality of life of the increasing aged population.
Conflicts of interest
The authors declare there is no conflict of interest.
6. Diabetes
SIRT1 is important for promoting glucose-stimulated insulin secretion in pancreatic β-cells 50, 51. Additionally, SIRT1 has a protective effect against insulin resistance in peripheral tissues, including adipose tissue, liver, and skeletal muscle 52. These findings suggest that SIRT1 is important for glucose homeostasis and the prevention of type 2 diabetes. Whole-body Sirt1-overexpressing transgenic mice, when fed a high-fat diet (HFD), have shown improvements in glucose tolerance through reduction of hepatic glucose production 52. Additionally, these mice do not show changes in body weight or composition. In the kidney of diabetic model mice, SIRT1 inhibits oxidative stress, which can lead to nephropathy, by induction of cyclooxygenase-2 (COX-2) expression 53. It has also been shown that administration of NMN ameliorates glucose intolerance in HFD-induced type 2 diabetic mice, enhances hepatic insulin sensitivity, and restores oxidative stress gene expression, and inflammatory responses, partly through the activation of SIRT1.
7. Non-alcoholic fatty liver disease
Non-alcoholic fatty liver disease (NAFLD) is characterized by steatosis of the liver and is linked with insulin resistance and metabolic syndrome. Studies have observed a reduction of sirtuins in NAFLD 54. SIRT1/3/5/6 are reported to be reduced in patients with NAFLD 54. This reduction is accompanied by an increase in lipogenic genes such as fatty acid synthase and SREBP-1. SIRT1 and SIRT3 have particularly been investigated in regard to NAFLD. SIRT1 expression is reduced by HFD 55. Overexpression of SIRT1 upregulates fatty acid oxidation pathways and downregulates lipogenic pathways, protecting the liver from steatosis. SIRT3 function is impaired in HFD, leading to hyperacetylation of target proteins in the mitochondria and impairing their activities 56– 58. SIRT3-deficient mice exacerbate these phenotypes, while overexpression can ameliorate NAFLD 59.
8. Atherosclerosis
SIRT1 has been shown to improve vascular function. SIRT1 is positioned to affect many pathways important for endothelial function 60– 63. SIRT1 suppresses the expression of inflammatory factors, including interleukin-6 (IL-6), monocyte chemoattractant protein 1 (MCP-1), intercellular adhesion molecule 1 (ICAM-1), matrix metalloproteinase 14 (MMP14), and vascular cell adhesion molecule 1 (VCAM-1) 64. Additionally, SIRT1 improves free fatty acid, triglyceride, total cholesterol, and blood glucose levels 65, 66. These protective effects of SIRT1 indicate that it acts as an anti-atherosclerosis agent. Consistent with these findings, NMN administration dramatically improves vascular function in aged mice, partly through the activation of SIRT1.
9. Alzheimer’s disease
Alzheimer’s disease (AD) is marked by multiple pathologies, including neuroinflammation, amyloid-beta plaques, mitochondrial damage, and increased oxidative stress 68, 69. Patients with AD have lowered expression of SIRT1 70, 71, which is recapitulated in the hippocampus of AD model mice 72– 75. SIRT1 activation is capable of reducing the amount of oligomerized amyloid beta through upregulating the production of alpha-secretase 68, 69. This is corroborated by mouse models overexpressing SIRT1 and amyloid precursor protein. Additionally, SIRT1 promotes neuronal function and survival in AD model mice. CA1-localized SIRT1 overexpression not only preserves learning and memory in AD mice but enhances cognitive function in non-AD model mice 76.
10. Retinal degeneration
Retinal degeneration is prominent in diseases such as macular degeneration and diabetic retinopathy. A recent study reported the importance of SIRT3 and SIRT5 in the survival of retinal photoreceptors 77. In particular, mitochondrial SIRT3 activity is sensitive to the reduction in NAD +. Decreases in retinal NAD +were detected in multiple retinal degenerative disorders, including age-associated dysfunction, diabetic retinopathy, and light-induced degeneration 77. Supplementation with the NAD + intermediate NMN was able to restore retinal function 77. These findings suggest a possible therapeutic treatment for a wide variety of diseases with photoreceptor degeneration.
11. Depression
Depression is a complex psychiatric disorder associated with a number of pathologies, including inflammation, synaptic dysfunction, metabolic syndrome, and cognitive deficit. Sirtuins have been shown to have a role in the development of depression 78. In the dentate gyrus region of the hippocampus, it has been shown that SIRT1 is decreased under conditions of chronic stress, which has been associated with depressive-like behaviors 79. Additionally, inhibition of SIRT1 by genetic or pharmacological methods has reproduced depressive behaviors. Activation of SIRT1 is able to lead to anti-depressive behaviors 79. However, it has been observed that SIRT1 regulates expression of monoamine oxidase A (MAO-A), which lowers serotonin and drives anxiety-like behaviors 80, indicating that a balance in SIRT1 expression/activity is important for mood disorders.
SIRT2 has also been reported in mood disorders. Hippocampal SIRT2 expression is decreased in chronic stress conditions 81. Pharmacological inhibition of SIRT2 recapitulates depressive behaviors. Adenovirus-mediated overexpression of SIRT2 produces anti-depressive behaviors, which were abolished when hippocampal neurogenesis was disrupted by X-irradiation 81.
Therapeutic potential of boosting NAD+ in aging and age-related diseases
Translational Medicine of Aging, (2018), Volume 2 PP. 30-37
- Johnson S, Imai SI.
NAD + biosynthesis, aging, and disease. F1000Res. 2018;7:132. Published 2018 Feb 1. doi:10.12688/f1000research.12120.1
FAQ
1. What is NAD+?NAD+ (Nicotinamide Adenine Dinucleotide) is a vital coenzyme in every cell, aiding energy production, DNA repair, and metabolism. Levels decline with age, so supplements like this 1000mg vial aim to restore them for better vitality and longevity. 2. Is NAD+ a peptide?No, NAD+ is a dinucleotide coenzyme, not a peptide. It consists of two nucleotides linked by phosphates, without amino acid chains or peptide bonds. This product is sold as a research-grade NAD+ compound.3. What are the benefits?NAD+ boosts mitochondrial function, ATP energy, sirtuin activation for anti-aging, and DNA repair. Users report improved energy, cognition, and recovery; it's studied for age-related issues like neurodegeneration.4. How to use NAD+ 1000mg?Reconstitute with bacteriostatic water, then inject subcutaneously (e.g., 50-100mg daily). Start low to assess tolerance; store lyophilized at room temperature, refrigerated after mixing. Research use only.5. Is it safe and what is purity?
High-purity (tested via HPLC) is intended for lab research, not for human use. Common questions indicate there are no major side effects at low doses, but it’s best to consult professionals. It is not suitable for pregnancy.
NAD+ 100mg
$48.00